
 扫描关注微信
扫描关注微信



得益于高能量密度和低成本,锂硫(Li-S)电池被认为是先进储能系统的有前途的候选者。尽管在抑制多硫化锂的长期穿梭效应方面做出了巨大努力,但对纳米级多硫化锂界面反应的理解仍然难以捉摸,这主要是因为原位表征工具在高时间空间分辨率下追踪不稳定的多硫化锂的液-固转化方面存在局限性,迫切需要了解Li-S电池内部的耦合现象,特别是多硫化锂的动态分布,聚集,沉积和溶解。
在此,厦门大学廖洪钢教授,美国阿贡国家实验室徐桂良教授和Khalil Amine教授等人通过使用高时空分辨电化学原位液相透射电子显微系统,直接在原子尺度上可视化了多硫化锂在电极表面上的转化。值得注意的是,在纳米团簇活性中心固定化表面上捕获了意外聚集诱导的多硫化锂聚集和电荷储存,它进一步诱导了多硫化锂的致密液相中非平衡Li2S纳米晶体的瞬时沉积。在没有活性位点介导的情况下,反应遵循经典的单分子途径,多硫化锂逐步转化为Li2S2和Li2S。分子动力学模拟表明,活性中心与多硫化锂之间的长程静电相互作用促进了由Li+和Sn2-(2
相关研究成果以“Visualizing interfacial collective reaction behaviour of Li-S batteries”为题发表在Nature上。

对此,郑州大学司玉冰副教授评价:这项工作填补了如何将高能量和低成本锂硫电池商业化的巨大知识空白。作者的成像结果解决了关于多硫化物穿梭的起源和演变以及这些电池中界面反应的缓慢动力学的长期争论,并证实了电极表面结构对这些过程的影响,这些结果对于电池和电子显微镜研究都具有重要意义。
Nature副主编Yohan Dall’Agnese高度评价:“这篇论文让我印象深刻,作者揭示了锂硫电池中一种完全出乎意料的电荷存储机制,考虑到这种类型的电池已经被广泛研究了几十年,这种情况极为罕见。由于开发了令人印象深刻的高分辨率原位透射电子显微镜,这些观察成为可能,这项工作将有助于改进下一代电池。”
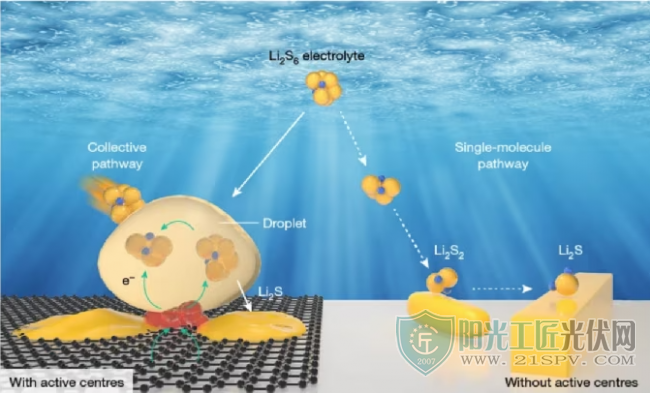
多硫化锂的聚集反应示意图
研究背景
锂硫(Li-S)电池作为一个经历了16电子的反应,将硫转化为一系列可变链长的锂多硫化物(LiPSs)。其中,四分之三的容量主要是由可溶性Li2S6与固体Li2S的反应所贡献的(图1a)。在此过程中,LiPSs的高溶解度和Li2S2/Li2S的绝缘性能会导致活性物质的持续损失和严重的容量衰减。其中,已经提出了许多策略来提高能量密度和循环稳定性,例如硫宿主的结构定制,隔膜设计,电解液工程等。然而,这些材料设计的基本原理仍然知之甚少。例如,阻碍快充锂硫电池发展的缓慢反应动力学的限速步骤是什么?活性中心如何保持活性以催化电极-电解质界面上的LiPSs?
为了研究Li-S电池的电化学反应,原位表征技术,包括X射线衍射(XRD)、X射线近边吸收结构谱(XANES)、核磁共振和拉曼光谱被使用。这些技术可以提供反应中间体/产物的特定化学/结构信息。然而,它们主要从电解液和电极中获取混合信号的统计信息,导致对LiPSs界面反应动力学的理解有限。得益于高时空分辨率,原位透射电子显微镜(TEM)可以在原子/单分子尺度上跟踪动态反应。目前,开孔构型几乎无法避免硫在高真空环境下的自发升华, 之前报道的液相电池结构是由电子束(e-beam)而不是电场驱动,在使用过程中不可避免地容易受到光束损坏,上述原位TEM研究尚未揭示液相电解液中的实际电化学氧化还原反应。
核心内容
在此,作者在液相电池内构建了Li-S纳米电池,并结合电化学透射电子显微镜(EC-TEM),实现了对基于醚类电解液中LiPS在电极表面的演变的高分辨率和实时观察(图1b-d)。研究表明,活性中心将可溶性LiPS聚集成液滴状致密相,并诱导非平衡纳米晶/非晶Li2S的瞬时沉积,而不是传统认为的分步转换。此外,密度泛函理论(DFT)计算和分子动力学(MD)模拟指出,聚集诱导的结晶是由LiPSs活性中心与液滴状稠密相之间的长程静电相互作用和集体电荷转移行为引起的。
EC-TEM的组装在安装EC-TEM支架上之前,作者组装了一个以Ti为电极的电化学液体电池,用于Li2S6电解液注入(图1c)。同时,为了提高液体电池的空间分辨率,应用了100 nm厚的隔膜和大约10 nm的SiNx观察窗口。在电化学液体电池中获得的循环伏安(CV)曲线展现了一对氧化还原峰(图1b),对应于LiPS的典型液-固转化。液体电池在−0.5 V下恒电位放电,从可溶性Li2Sn(Li2Sn+e-→Li2S,22S。透射电镜图像显示了两种不同形式的Li2S沉积-超小纳米晶(有活性中心)和棒状/板状结构(无活性中心),表明电极表面与Li2S沉积的热力学/动力学之间有很强的相关性。
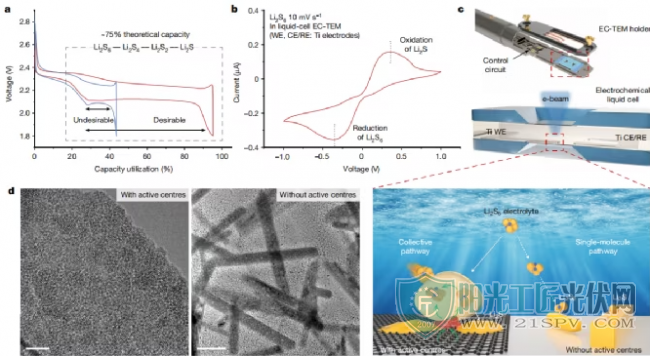
设计的液相EC-TEM来研究LiPSs的界面反应
通过高分辨率原位EC-TEM研究研究了不同电极表面的电化学反应,透射电镜图像和投影面积变化揭示了在没有活性中心的情况下Li2S沉积/溶解的的形貌演变,无活性中心介导(图2a、b)。在−0.5 V的恒电位放电过程中,观察到两种类型的成核-生长途径:一种是棒状/板状Li2S的单步沉积,另一个是通过亚稳Li2S2的两步沉积。对于两步途径,在初始成核过程中在电极-电解质界面处形成固体纳米核,作为随后生长(25 s)的种子。约70s后,初级Li2S2粒子的生长速度减慢。然后,亚稳态迅速溶解,而颗粒沿其纵向方向拉长(116.2 s)。在颗粒2完全溶解和质量再分配后,形成了棒状Li2S(137.6s)。在静电荷(+0.5 V)过程中,棒状Li2S逐渐缩短(201.6 s),然后迅速分解(219.8 s)。由于Li2S的绝缘性能,溶解速率的变化(约240s前后)在初始阶段产生了较大的能垒。此外,棒状Li2S也可以通过单步途径直接沉积,还发现还原的Sn2-(n≤2)离子具有可扩散的性质,导致固体Li2Sn(n≤2)在电极表面附近沉淀。
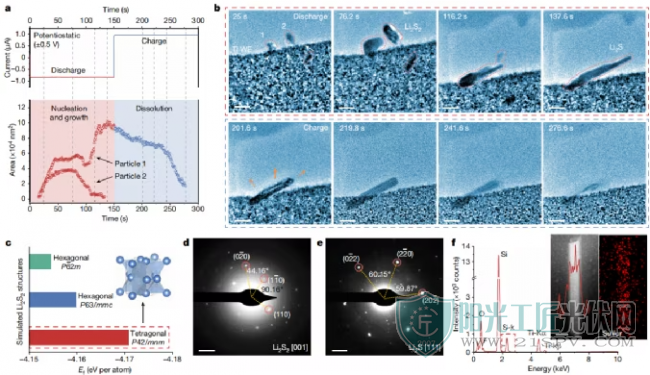
无活性中心的LIPSs的界面反应和结构
聚集路径为了研究金属活性中心在LiPSs氧化还原反应中的作用,合理设计了Mo纳米团簇/N掺杂石墨烯(Mo NCs/N-G)。在-0.5 V的恒电位放电期间,棒状Li2S没有成核生长,而Mo NCs/N-G纳米片则逐渐扩散(185 s)。随后在更高分辨率下的观察显示,在放电过程中,石墨烯的边缘和表面形成了许多暗区,对应于Li2S沉积在Mo NCs/N-G纳米片上。与裸电极表面和位于左下角没有电子接触的Mo NCs/N-G纳米片相比,由于Mo NCs/N-G纳米片上的红色虚线框突出显示,观察到明显的对比变化。
在液相电池中Mo NCs/N-G纳米片上电化学沉积后,在原子尺度上对Li2S的结构进行了研究。高分辨率透射电子显微镜(HRTEM)图像显示,Li2S以非平衡晶体形式沉积在Mo NCs/N-G纳米片上(图3e),沉积区域由纳米晶和非晶Li2S组成(图3f),而沉积在裸电极表面的为单晶面中心立方Li2S。此外,通过SAED图案的多晶衍射环进一步证实了这一点。
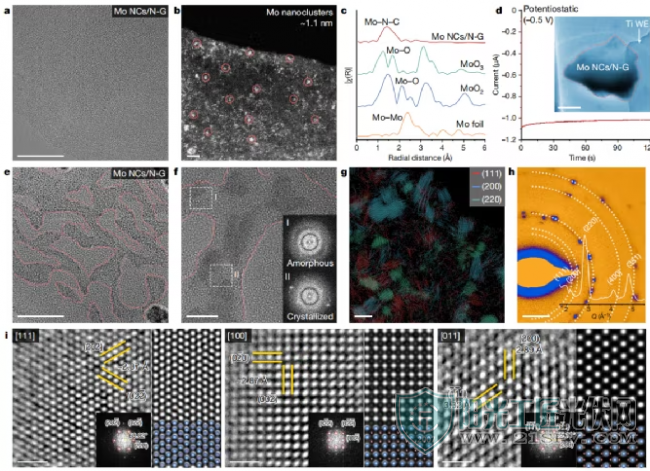
有活性中心的沉积Li2S的结构
在±0.5V的恒电位条件下,进一步研究了活性中心介导的LiPSs的界面反应和可逆性。
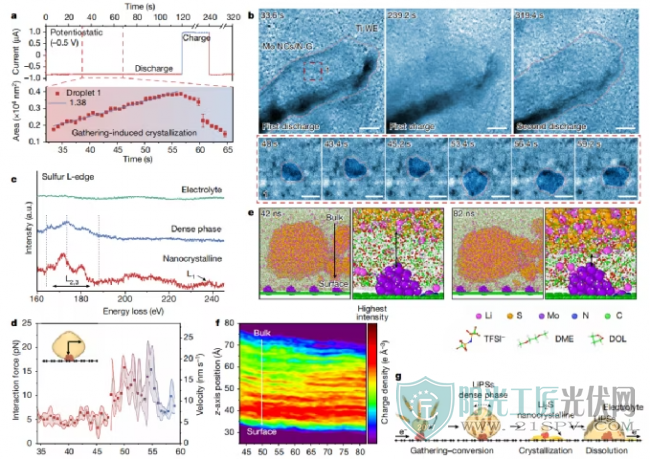
由活性中心介导的LiPSs的界面氧化还原反应
结论展望
综上所述,在Li-S电池的多电子反应过程中,涉及LiPSs中间体与活性中心之间的吸附、催化和转化,以及控制Li2S沉积/溶解的反应动力学的复杂性导致了多种多样且模棱两可的途径。作者通过使用高时空分辨率原位EC-TEM,表明活性中心将可溶性LiPS聚集成液滴状致密相,从而诱导瞬时结晶,而不是经典的逐步转化。此外,初步结果表明,聚集反应机制似乎对其他金属活性中心具有普遍性,需要更系统的研究。以往对材料/表面改性的研究主要集中在从单分子方面阐明吸附和催化机理。实验和模拟结果表明,离子和分子的聚集状态和集体行为在电化学界面反应的动力学中起着关键作用,该聚集机制为构建下一代高能量、长寿命和快速充电的锂硫电池提供了新的见解。
论文信息
发布期刊 Nature
发布时间 2023年9月6日
文章标题Visualizing interfacial collective reaction behaviour of Li–S batteries
原标题:厦门大学廖洪钢团队:审稿意见长达100页,打开锂硫电池“黑匣子”!



 宁德时代吴凯...
宁德时代吴凯... 天合光能陈奕...
天合光能陈奕... 刘岩: 追光行...
刘岩: 追光行... 黄震院士:大...
黄震院士:大...